Chapter 2
Timely access to new cancer medicines
2.1
A key theme throughout the inquiry has been the need for a fundamental
review of the regulatory and reimbursement processes for cancer drugs. Some
submitters told the committee that the current process has served Australia
well, but expressed concern that without 'modernisation' it would not be able
to keep pace with the growing trend in applications for new medicines.[1] Some
submitters described the current process as complex and time-consuming and
considered that it delivered suboptimal outcomes for Australian patients.[2]
2.2
Chapters 2 and 3 of this report examine the regulatory pathway for
subsidised access to new cancer medicines in Australia and consider some
aspects of that pathway that may be contributing to delays in access. Broadly
speaking, evidence to the inquiry identified four key areas of concern:
timeliness of decision making; assessment of cost-effectiveness; the need for
greater consumer input and improved access to information. This chapter will focus
on the first of these factors, while Chapter 3 will consider the assessment of
cost-effectiveness, the impact of delayed access on cancer patients and the
role for greater consumer input.
Timelines for access to new medicines in Australia
2.3
As noted in chapter 1, the committee heard that one of the key factors
affecting access to medicines is the timing of applications by pharmaceutical
companies to the TGA seeking registration of medicines and to the PBAC seeking
reimbursement. The Department of Health (DOH) noted that for cancer medicines
submitted for TGA approval between 2009-2014, submissions were made an average
of 38 weeks after the lodgement of a submission to the United States (US) Food
and Drug Administration (FDA) and an average of 38 weeks after the lodgement of
a submission to the European Medicines Agency (EMA). DOH told the committee
that this approach is often a function of the size of the Australian market:
This kind of business
approach seeks to establish, as early as possible, a positive response in the
regions offering the most potential for profit, due to their large population
size. This avoids the situation where a deferral or rejection from a country
with a small population, like Australia, could influence other authorities,
thereby jeopardising the profit margins that could be achieved in larger
countries/regions.[3]
2.4
The committee notes that this factor is outside the control of the TGA
and PBAC.[4]
DOH told the committee:
The ability to
deliver timely access to medicines is also affected by the timing of the
applications which, in Australia, is at the discretion of pharmaceutical
companies. It is acknowledged that these companies operate in a global industry
and this can affect their decisions. Sponsors often choose to apply first in
the US or Europe, delaying consideration of the medicine in Australia.[5]
2.5
The PBAC said that comparisons of dates of regulatory submissions show
that new products are submitted to the TGA 'a median of 105 days after they are
submitted to the EMA, although the TGA accepts the same evidence package as the
EMA'.[6]
2.6
In contrast, pharmaceutical companies provided some positive examples of
the introduction of new medicines into the Australian market. For example, Bristol-Myers
Squibb submitted:
... Australia was the
second country in the world to approve ipilimumab (known as YERVOY) for the
treatment of patients with advanced or metastatic melanoma in June 2011, just
three months after its approval by the Food and Drug Administration (FDA) in
the United States.[7]
2.7
The committee notes that this timely approval of ipilimumab may reflect
the fact that Australia has the highest incidence of melanoma in the world.[8]
TGA registration process
2.8
The TGA registration process consists of eight phases with established
timeframes specified under the Therapeutic Goods Act 1989.[9] The committee
received evidence suggesting that there is merit in reviewing the registration
process to identify circumstances in which a more flexible approach might be
supported. In particular, submitters identified options through which Australia
might leverage off overseas regulators or learn from their experience.
2.9
As noted earlier, the committee heard that it is rare for international
sponsors to seek registration in the Australian market ahead of applications to
the FDA or the EMA. Submitters noted that the TGA accepts the same evidence
package as the EMA and proposed that the TGA registration process could be streamlined
by taking account of circumstances where a medicine had been assessed and
approved by a recognised regulatory body.[10]
2.10
Some submitters suggested automatic conditional approval for drugs
approved by the FDA or EMA.
2.11
Cancer Action Victoria noted the recommendation of the National
Commission of Audit that recognising approvals make by overseas authorities
would provide better outcomes for consumers by cutting delays caused by the
approval process and reducing the estimated administrative costs incurred by
pharmaceutical companies.[11]
2.12
Similarly, Merck Sharp and Dohme (MSD) proposed that the TGA could adopt
the evaluation reports from an overseas regulatory authority as the basis of an
Australian approval:
Evaluation reports from an approved regulatory authority
would be assessed before an independent sovereign decision was made by the
TGA. Through leveraging international experiences and resources, patients in
Australia could secure timelier access to medicines, whilst the TGA would make
significant efficiency gains by reducing duplication of effort.
2.13
MSD suggested that by adopting this approach, new medicines could be
made available to Australian cancer patients in as little as three months after
overseas approval.[12]
2.14
Leukaemia Foundation of Australia told the committee:
We would support for rare cancers in particular where drugs
are approved in the United States and Europe that there be an automatic
conditional acceptance through the TGA of those drugs and bringing a much
shorter time for those drugs to be available. We also support a managed access
scheme through the PBS to bring those drugs in for those rare cancers.[13]
2.15
However Cancer Voice SA cautioned against adoption of overseas approvals
without first undertaking an evaluation of past data to demonstrate that such
an approach would produce better, different or faster decisions.[14]
2.16
Submitters also noted that Australia has no process in place to expedite
the review of critical or breakthrough medicines.[15]
Cancer Council Australia and Clinical Oncology Society of Australia (CCA/COSA)
told the committee:
The [EMA] in the United Kingdom and the [FDA] in the United
States provide the opportunity for expedited approval in a shorter timeframe
and in some cases based on earlier indicators of effectiveness, for
breakthrough therapies. The EMA and FDA regulators allow companies to test
cancer drugs using surrogate measures instead of overall survival and other
patient centred measures such as tumour size and progression depending on the
medicines fit for purpose. [16]
2.17
In its submission, MSD provided an overview of the FDA's Fast Track
Designation and Breakthrough Therapy Designation. Key features of both
designations are the level of interaction between the FDA and the sponsor and
the degree of flexibility with respect to the submission of the marketing
application. In both models early and frequent interaction ensures marketing
applications and compressed development programs still meet the FDA's rigorous
standards for safety and effectiveness while facilitating earlier access to
important medicines for cancer patients.[17]
2.18
DOH told the committee that, while there is currently no formal
expedited evaluation system, if the TGA considers an application to be a
significant therapeutic advance or of critical importance, it will, 'wherever
possible, work with the relevant applicant with a view to facilitate an early
decision, provided the product meets the TGA's quality, safety and efficacy
requirements.'[18]
2.19
Mr Christian Sellars, Merck Sharp and Dohme, outlined a case study for
the committee to illustrate the difficulties MSD perceives in the TGA
registration process:
I thought I might, if you will indulge me, tell the brief
story of one research area that was mentioned ... by the Australian Melanoma
Research Foundation, some of your earlier witnesses. This is the type of
product that Ron Walker, the Melbourne businessman, was treated with. The area
is called immunotherapy and it is a very promising new area of research.
MSD was very fortunate over the weekend to publish our first
head-to-head trial in the New England Journal of Medicine on melanoma for this
cancer area. What we are seeing is that about a third of patients are seeing a
visible reduction in their tumours. That reduction is sustained over a year,
which is a fantastic outcome in a cancer type that has resisted effective
treatment for many years. What is probably most exciting about this area—and it
is not just MSD; there are four or five companies that are investing
substantial amounts in trials. Our organisation alone has 150 trials in this space
on 30 different tumour types. It is probably taking up the biggest chunk of our
$7 billion research budget at the moment. But it is showing substantial value
across many of the cancer types that we have tried it in so far.
So, when we still had phase 1 data only, which was last year,
we put a submission in to the TGA. The TGA has no formal fast-track approval,
but we felt that the importance of this therapy in an area of very severe need
justified a not-normal approach, so we put in based on phase 1 data and we were
very fortunate that the TGA processed us and the approval came through within
eight months from submission. The normal approval time is 12. That was approved
on Friday.[19]
2.20
Mr Sellars told the committee that, rather than an example of the system
working, the process of submitting this product illustrated the uncertainty
that companies face when deciding to seek early approval for a treatment:
The process of submitting this product has actually been
extraordinarily disruptive and ad hoc. We have had no certainty about how data
would have been considered, when decisions would have been made, how the TGA
process would line up to the PBAC process and whether we would find ourselves
in a situation where these cogs did not quite line up perfectly and,
effectively, we would lose our slot.[20]
2.21
The committee heard that a further impediment to timely access to cancer
medicines is the requirement for an application for the registration of a new
indication for a medicine already listed in the ARTG, to be made by the sponsor
of medicine.[21]
Medical Oncology Group of Australia (MOGA) told the committee that the PBS
currently has inadequate coverage of new indications that are outside the
TGA-approved indications, despite the availability of evidence to support the
new indication. The committee heard that there are a number of reasons the
registration of indications with the TGA does not keep pace with evidence
development including:
-
the complexity of the approval process;
-
only drug sponsors are permitted to lodge an application for a
new indication;
-
a lack of commercial incentives for the sponsor to seek further
approval; and
-
data ownership issues in circumstances where evidence may be
developed by research institutions without the involvement of the original
sponsor.[22]
2.22
The committee heard that addressing these issues, to allow clinicians
and/or patient groups to lodge an application for a new indication for an
already registered medicine, could improve the responsiveness of the registration
process to changes in the clinical setting.[23]
2.23
The committee notes the independent Review of Medicines and Medical
Devices Regulation, announced in October 2014, has examined the regulatory
framework administered by the TGA. The review has sought to identify:
-
areas of unnecessary, duplicative, or ineffective regulation that
could be removed or streamlined without undermining the safety or quality of
therapeutic goods available in Australia; and
-
opportunities to enhance the regulatory framework so that
Australia continues to be well positioned to respond effectively to global
trends in the development, manufacture, marketing and regulation of therapeutic
goods.
2.24
The independent Expert Panel (Panel) provided the Government with its
first report on 31 March 2015 and the committee notes that the Panel's report
includes recommendations to:
-
expand the pathways by which sponsors can seek marketing approval
for a medicine or medical device, including making provision for utilisation of
assessments conducted by comparable overseas regulators, and for expedited
assessments in defined circumstances; and
-
enhance transparency and predictability of processes and
decisions to build trust and confidence in the TGA's ability to ensure
Australians have timely access to high quality, safe and efficacious products.[24]
Time to PBS listing
2.25
The committee heard that, from a cancer patient's perspective, the
critical timeline is that between regulatory approval of a cancer medicine by
the TGA and its listing on the PBS. Submitters spoke of a significant time lag
between these two regulatory decision points.[25]
The committee heard varying estimates of the average time of this lag.
2.26
Medicines Australia (MA) submitted that the average time from
registration of a medicine by the TGA to reimbursed access on the PBS is in
excess of 18 months:
-
new listings take on average 589 days (over 1 ½ years), compared
to 456 days in Canada, 584 in England and 256 in France; and
-
subsequent listings take on average 700 days (nearly 2 years),
compared to 189 days in Canada, 474 in England and 365 in France.[26]
2.27
MA said that 'disturbingly, some medicines took up to 1,600 days (4½
years) for a new listing and 2,400 days (more than 6½ years) for a subsequent
listing'.[27]
At the committee's hearing, MA stated that new cancer medicines can take six
months longer than other types of medicines—on average 1.6 years from TGA
registration to PBS listing.[28]
2.28
Novartis Oncology Australia New Zealand (Novartis) told the committee:
In research
specifically conducted for our submission, we found when reviewing listings for
all medicines since 2011 that it takes an average of almost 2½ years—890
days—for a cancer medicine to be listed on the PBS from when it was approved by
the TGA. During the same time period non-cancer medicines are taking less than
half as many days—391 days.[29]
2.29
DOH provided indicative timelines for each phase of the regulatory and
reimbursement process. Figure 2.1 below indicates that the expected time
between regulatory approval by the TGA and PBS listing can range from between
seven to 18 months.
Figure 2.1: Pathway for Access to New medicines in Australia
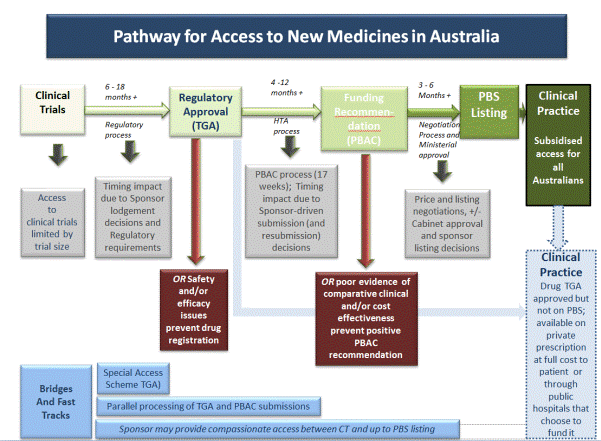
Department
of Health, Submission 197, p. 10.
The PBAC assessment process
2.30
The time for the PBAC review of an application for a medicine is
currently 17 weeks from submission of the application to recommendation to the
Minister.[30]
DOH told the committee that the PBAC has one of the fastest reimbursement
processes in the world.[31]
2.31
DOH provided an overview of the PBAC process, highlighting the rigorous
and formal nature of the process and the volume and complexity of the analysis
undertaken during the 17 week timeframe.
There are two aspects to the process. First of all, the
process is rigorous and it is formal. There is a substantive amount of evidence
to go through. There is complex work, and there is a large volume of work that
is all written. That is the purpose of the application. It is a written
application. That application is then assessed. The department contacts six
academic institutions around the country. Those groups are specialist health
technology assessors. They produce a written and comprehensive evaluation
report. That evaluation report is provided to the company. The company has the
opportunity to respond. That report, together with the comments and feedback
from the company, goes in front of two of the subcommittees of the PBAC: the
economics subcommittee and the drug utilisation subcommittee. After the
subcommittee meetings, those subcommittees issue their advice to the PBAC. That
advice is also provided to the company, and once again the company has the
opportunity to respond to that. All that material is provided to the PBAC
within the 17-week cycle.[32]
2.32
However, MA submitted that very few applications receive a positive
recommendation in 17 weeks from the lodgement of the first submission and
identified this as a key factor in the time lag from TGA approval to PBS
listing.
Most medicines and applications for new indications require
more than one submission to achieve a positive PBAC recommendation and
subsequent PBS listing. To prepare and resubmit, following an initial
rejection, takes at least one and sometimes more cycle(s) such that it commonly
takes 12-18 months for a positive decision, and can take several years.[33]
2.33
MA told the committee that the average number of submissions required to
obtain a positive recommendation from the PBAC for cancer medicines is 2.3 for
new listings, equating to approximately three years, and 2.5 for subsequent
listing (equating to approximately 3.5 years).[34]
2.34
Submitters argued that the fixed, cyclical nature of the PBAC's
assessment process means that if an application for listing is unsuccessful and
the sponsor needs to resubmit, this can add substantially to the timeline.
Novartis told the committee:
It is often the case for cancer medicines that the submission
may be rejected or deferred by the PBAC leaving the sponsor to wait until the
next available PBAC cycle to resubmit. This is leading to sponsors and the PBAC
playing out a long drawn out negotiation using multiple submissions and
multiple PBAC meetings. This often takes years and uses significant resources
on both sides.[35]
2.35
Submitters also noted that preparation for a major submission to the
PBAC takes, on average four to six months and is costly and resource intensive.[36]
As noted above, the tendency for applications to require resubmission to
address new issues, to provide more complete data or to change the type of
analysis can add significantly to the timeline for PBS listing.
2.36
Witnesses suggested that alternative processes should be explored to
expedite the resubmission process. Some witnesses suggested that a designated
fast-track approval process should be implemented, while others suggested
adopting a tiered submission process has the potential to allocate resources
according to need, by freeing up resources from less complex applications to
allow greater focus on more complex, high-risk medicines, or those with a
higher clinical need.[37]
Submitters noted that similar approaches have been implemented in the US and in
the Netherlands.[38]
2.37
DOH stressed that the TGA and the PBAC ' are very keen to be adaptive
and flexible where there is a need to'. Ms Felicity McNeill, First Assistant
Secretary, noted:
We had some cancer drugs at the July meeting that were not initially
given positive recommendations. We were then able to bring together stakeholder
groups such as consumers and clinicians to do some work and bring it back to
the November PBAC meeting, where it got a positive recommendation.[39]
2.38
The committee heard that a significant factor in delays in securing
listing approval stems from difficulties associated with assembling trial data
that is sufficiently robust to satisfy PBAC requirements early in the
assessment process.
2.39
Both DOH and PBAC submitted a key factor influencing both the timing of
PBAC decisions and the likelihood of a successful application for listing is
the quality of evidence provided to support the application.[40]
There is an increasing trend for the clinical evidence
documenting the effectiveness and cost-effectiveness of new cancer medicines to
be of such poor quality that it does not allow confident assessment of benefit.
For example, studies without proper comparison groups are increasingly being
used as the basis of proposals for listing. Even when well-designed comparative
trials are conducted the data presented are often from early analyses. Decades
of research have consistently shown this type of data will over-estimate the
benefits of a new medicine or other intervention.[41]
2.40
Novartis suggested that consideration could be given to elements of the
assessment approach adopted in the US, which accepts earlier data and employs a
system of rolling submissions which allows sponsors to submit additional data
as trials progress and data emerges.[42]
2.41
The committee heard that, while the PBAC undertakes its evaluation
largely on the basis of the information provided in the applications, there is
some flexibility in the current system to enable a sponsor to provide
additional information during the assessment period in certain circumstances.
The former Chair of the PBAC stated:
The PBAC works with the evidence that is presented in the
sponsored submission. We do not invent new data or go and find new data. The
evaluation process, at least, provides the sponsor along the course of the
17-week pathway at the moment the opportunity to see the evaluation and the
appraisal, and any additional analyses are done. That is very much based on
what the sponsor actually submits. Should the sponsor, for example, submit to us
as part of the parallel process with an unspecified patient population, because
TGA has not yet finalised the patient population that is most suitable for the
drug, and we along the way get the TGA's proposed patient population, then the
evaluation process can provide the sponsor the opportunity to resubmit some
limited data in the 17 weeks that exists currently to, shall we say, revise the
analysis or refine the analysis to match what is coming through TGA.[43]
Pre-submission planning meetings
2.42
Submitters suggested that one means of addressing the 'churn' in the
application process and improving the likelihood of successful applications
would be to provide for pre-submission planning meetings. MA told the committee
that sponsors would welcome the opportunity to meet with evaluators early in
the application process 'to provide clarity around the evidence, the form of
submission, the data required, [and] the appropriate pathway':
We would welcome a more open and more engaging, a more, if
you like, user-friendly process in which a sponsor and the government and
perhaps, ideally, the evaluator, can come together early, understand issues,
challenges and opportunities and see the way clear to provide as much
appropriate certainty and clarity as can be.[44]
2.43
The committee heard that, while such meetings may be arranged at the
request of a stakeholder, submitters see value in formally incorporating such
meetings into the assessment process.[45]
2.44
The committee notes that the assessment system applied by the National
Institute for Health and Care Excellence (NICE) in the United Kingdom, includes
early scoping meetings to discuss the 'decision problem', secure agreement on
the comparator and on the appropriate endpoint for determination of
cost-effectiveness prior to the sponsor making a submission.[46]
Similarly, pre-submission planning meetings are a feature of the system
administered by the pan-Canadian Oncology Drug Review (pCODR). The pCODR
pre-submission process takes place between six to 12 months prior to the
lodgement of the submission and aims to assist the submitter and other
stakeholders through the process.[47]
2.45
In its White Paper, Improving Access to Cancer Medicines, the
Cancer Drugs Alliance notes that there is significant value in improving early
multi-stakeholder engagement, including: improved understanding of the drug and
disease area in advance of initiating the submission and evaluation,
improvements in the relevance and consistency of the assessment process and
identifying important factors for inclusion in the application.[48]
2.46
By contrast, while acknowledging that there is always room for
improvement, DOH told the committee that the existing process is 'fundamentally
based on constant engagement with pharmaceutical companies'. DOH illustrated
this by describing the process applied to the assessment of the drug
pembrolizumanb. Ms Adrian Platona, Assistant Secretary, Pharmaceutical
Evaluation Branch said:
For that particular drug, which has received a lot of
attention today and in the media recently, we, the department had a least three
meetings with the company to discuss the nature of the application and the
evidence in the application.[49]
Parallel processing
2.47
A number of submitters told the committee that a key factor in delays in
listing of medicines on the PBS is that application to the key regulatory
bodies, the TGA, PBS and Medical Services Advisory Committee (MSAC)[50]
is sequential and dependent on fixed meeting dates.[51]
2.48
Since January 2011 sponsors have had the option of progressing
applications through the TGA and PBAC processes simultaneously.[52]
Theoretically, submissions assessed via the parallel process will have
compressed timeframes.
2.49
However, the committee heard that while assessment by the TGA and PBAC
happens in parallel, the process does not necessarily result in faster listing
of medicines.[53]
DOH advised the committee that to date 20 per cent of major applications for
cancer medicines have used this option.[54]
MA told the committee that between 2011 and 2014, 27 per cent of major
submissions to the PBAC had used the parallel TGA-PBAC process and indicated an
expectation that the figure will grow.[55]
MA subsequently noted that the time to listing 'varies greatly' regardless of
whether drugs are assessed via the parallel process.[56]
Novartis indicated that the average time from TGA approval to PBS listing has
increased since 2011 when the parallel process was introduced. Data
commissioned by Novartis showed that for oncological drugs, the mean time for
approval to listing was 637 days prior to July 2011, and increased to 890 days
after July 2011.[57]
2.50
Submitters noted that parallel processing is a relatively new process
and suggested that there is some scope for fine tuning. MA told the committee
that there has been a lower recommendation rate for cancer medicine submissions
using parallel processing, and noted that there is no guarantee that parallel
processing will result in a faster listing.[58]
2.51
Ms McNeill, First Assistant Secretary, Pharmaceutical Benefits Division,
DOH, expressed disappointment at the concerns raised regarding parallel
processing.
We have seen some really fantastic outcomes in that space
and, yes, we have seen some that have not been successful at all. There is a
bit of risk management there and we do accept that. When you are going to the
TGA and you are not quite sure what the final registration may be and you are
looking at the subsidy for that particular indication, we appreciate there is a
risk with that. But there also becomes an opportunity with that too, and that
is why we have engaged in this process with industry to try and further that.
We always learn from these things; systems evolve. But if 30-plus percent of
submissions are coming through this, there must be something in there that is
going reasonably well.[59]
2.52
In its submission the PBAC stated that the decision of some sponsors to
submit applications well in advance of TGA approval may be distorting the time
to approval:
Provided the data package is adequate and the price requested
by the sponsor is reasonable and found to be cost effective, the PBAC may be of
a mind to recommend approval before the final approval by TGA (e.g. dabrafenib
for melanoma). However, some sponsors are now choosing to submit applications
to the PBAC so far in advance of TGA approval that the PBAC has no option but
to reject or defer them, as the TGA-approved indication is critical to
determining a PBAC listing. This practice may be distorting the reported time
to approval.[60]
2.53
Roche Products's evidence confirmed that, in deciding whether to submit
an early application for parallel processing, sponsors consider the likelihood
of satisfying PBAC data requirements on the basis of the data available.
[W]e certainly aim to submit our applications under parallel
process or at the earliest opportunity. Where there have been delays or
decisions not to submit at that earliest opportunity or through parallel
processes, because of the PBAC's need for data certainty, our company may
decide to delay that application until additional data become availabile to
minimise that uncertainty or to conduct additional assessments to identify the
population where the drug is most cost-effective.[61]
2.54
Submitters told the committee that greater collaboration is needed between
each of the regulatory and reimbursement agencies with regard to the assessment
of clinical evidence to enhance the efficiency of parallel processing.[62]
The committee notes that the Review of Medicines and Medical Devices Regulation
recognised the synergies between the TGA, the PBAC and the MSAC and considered
that there would be benefits in considering organisational structures to
facilitate improved integration of these functions across the lifecycle of
medicines and medical devices.[63]
2.55
The committee notes that additional timing complexities are associated
with the assessment of co-dependent technologies, and that there is a view that
systems improvements have failed to address these:
It is common for cancer medicines, particularly targeted
medicines, to have an associated diagnostic test or treatment-associated device
to ensure the medicine is used where most effective.
Submissions for targeted medicines partnered with a
diagnostic test are complex in terms of content and process. They currently
require a separate recommendation from two separate committees with differing
meeting schedules; the Medical Services Advisory Committee (MSAC) for the test
and the PBAC for the drug. There appears inadequate interaction between the two
committees, and the submission processes vary greatly between the two.[64]
Timely and transparent price
negotiations
2.56
The committee heard that another source of delay in the listing of
cancer medicines is the post-PBAC negotiations between the sponsor and government
over price. The PBAC submitted that:
Delays following a positive recommendation by PBAC may be due
to inability of the sponsor and Government to agree on the price and other
details of financial agreements. For example, there was an 18-month delay
between the Committee’s recommendation for the listing of abiraterone for metastatic
prostate cancer and the sponsor agreeing to supply the drug on the PBS under the
recommended circumstances. During this period, there were multiple additional
applications for the same product and listing that had to be reviewed by the
PBAC.[65]
2.57
DOH clarified that while a decision not to proceed with listing may
reflect the sponsor's dissatisfaction with the PBS subsidy, it may also reflect
a desire to seek changes in the approved indication for the drug. Ms McNeill, said:
It can sometimes be both, but more often than not it is about
price, that is usually the vast majority of the concerns we have.
...
Often you are struggling as a pharmaceutical company to
demonstrate the value of your drug in the order of when you may be used in a
treatment cycle—whether you are first line, second line or third line.[66]
2.58
Ms McNeill further explained:
When you have a positive PBAC recommendation it is not like
you can never come back and ask for that to be changed. But other drug
companies will often put up their drug, take the recommendation and list on the
PBS so that the patient has subsidised access from the word go; and then they
put in resubmissions to the PBAC to seek changes in indication or changes in
price thereafter. It is entirely up to a drug company which way they choose to
do it. In [the case of abiraterone], they chose not to list and then continued
to argue. They decided not to go for the PBS subsidy but to leave it in the
private market until they got the recommendation they wanted.[67]
2.59
Novartis recommended that a negotiation period should be established for
all parties:
Once a cancer medicine has received a positive PBAC
recommendation, and an opportunity exists to list the medicine, a negotiation
framework and prescribed timeline (6 months) should implemented to ensure all
parties (i.e. DoH, PBAC and Sponsor) may reach a timely outcome.[68]
2.60
CanTeen submitted that there is need for greater transparency in the
pricing of cancer drugs with particular reference to utilising the clinical evidence
to increase the alignment between the price of cancer medicines and their
effectiveness.[69]
2.61
PBAC also expressed concern that public discussion of new cancer
medicines does not pay sufficient attention to the benefits and harms, as well
as the cost of new medicines, stating that:
It is highly likely that earlier access to cancer drugs will greatly
increase cost to the community if the mechanism by which earlier access is
granted involves acceptance of prices that result in much higher estimates of
cost-effectiveness.[70]
Transparency
2.62
A number of submitters commented on the need for greater transparency
throughout the TGA and PBAC process. Dr Katherine Nielsen, Director of Research
and Advocacy, Leukaemia Foundation of Australia told the committee that greater
transparency could lead to greater procedural efficiency and would help the
public to understand the reasons for delays:
We have seen in many submissions that it averaged 31 months
in 2012 for cancer drugs and generally required more than one submission.
Whether this is due to price expectations or unrealistic requirements in
demonstrating clinical effectiveness is not clear because the processes are not
actually transparent, so we do not really know.[71]
There is a lack of transparency about how decisions are made
at the PBAC level and the MSAC level. It would be good to have better
transparency for the public and also better expectations between the
parties—the sponsor and the government—in terms of what is needed to
demonstrate the value and cost-effectiveness of a drug and how to improve that.
But we also need to have transparency around that so that people understand why
there are delays. At the moment, we do not know why there are delays; there
simply are.[72]
2.63
The PBAC also recommended an increase in transparency around committee
processes, particularly the evidence provide to the PBAC. Noting that some
'high value' commercial information may need to be withheld, PBAC stated that
the majority of documents submitted to the PBAC can, and should be made
publicly available.[73]
Dr Suzanne Hill, Former Chair, PBAC, explained this position at the committee's
hearing:
We believe that there should be an agreement between
industry, the government, patients and physicians to have much more of the
submitted documentation released to the public. Likewise, we believe that as
much as possible of the documentation that is generated during the evaluation
process should be made available. We are concerned that without such a change
there will continue to be the fundamental misconceptions about the committee's decision
making that have emerged in some of the submissions to this inquiry. More
importantly, there will continue to be misinterpretation of data in and by the
media, and patients will continue to be under pressure to obtain access to
medicines that really may not offer them any value at their own sometimes
considerable expense.[74]
2.64
MA rejected these claims stating that the current transparency processes
in Australia are among the best in the world and have resulted 'from extensive
dialogue between industry and Government about the best way to implement the
process,' while still respecting legitimate commercial in confidence
considerations.[75]
2.65
Submitters noted that greater transparency would assist sponsors and
consumer groups to identify the reasons why applications have been unsuccessful
and help to reduce submission 'churn'.
2.66
The Unicorn Foundation told the committee:
We put so much effort into these submissions but do not
actually find out why it has been rejected or why not. I think that it is even
just opening the lines of communication with consumers and consumer groups on
how to make effective submissions but also on why a drug was passed or why not,
based on the actual evidence put forward.[76]
2.67
Witnesses noted that greater transparency would also assist patients and
their oncologists to make informed choices about treatments.[77]
Committee view
2.68
The committee notes that the processes for assessing applications for
registration and listing are appropriately rigorous and are based on clear
cyclical timelines. At the same time the committee notes the concerns raised by
sponsors and other stakeholders regarding the potential for inefficiency and
uncertainty in the system.
2.69
The committee considers that Australia should strive to achieve world's
best practice in the approval of medicines and should therefore maintain a
commitment to continuous improvement of its assessment processes. The committee
also notes that the pharmaceutical industry has a significant role to play in
achieving timely listing of cancer medicines.
2.70
The committee has received evidence pointing to fast track processes
used by overseas regulators and notes that key features of such processes are
early and frequent interactions between the regulator and the sponsor and a
process of 'rolling review'. These mechanisms ensure collaboration in the
design of trials to collect data that will support registration together with
the flexibility to submit sections of the application for review as they are ready.[78]
2.71
The committee considers that some of the suggested avenues for
streamlining the assessment process, particularly in the case of resubmitted
applications, merit further consideration, for example:
-
pre-application planning meetings to assist sponsors and other
stakeholders to better tailor their applications to the requirements of the
PBAC;
-
the scope for a tiered assessment process that matches resources
to the complexity of applications; and
-
a review of the parallel processing arrangements to identify opportunities
to allow flexibility in the submission of data in order to achieve compressed
timeframes in appropriate circumstances.
2.72
The committee notes concerns raised regarding the timeliness and
transparency of pricing decisions and notes that the Review of Medicines and
Medical Devices Regulation made similar findings. The Review has made
recommendations to improve transparency and predictability of TGA processes.
The committee considers that greater transparency throughout the TGA and PBAC
processes would aid understanding of the requirements of the assessment process
and would support cancer patients and their oncologists to make informed
choices with regard to their treatment. Greater transparency would also help to
dispel any misconceptions regarding the assessment of particular medicines.
Navigation: Previous Page | Contents | Next Page