Chapter 2 - Hepatitis C in Australia
2.1
This Chapter provides a brief overview of hepatitis and the
understanding of blood and blood safety in developed countries, paying
particular attention to improvements in diagnostic technology in relation to
hepatitis C. It also examines Australia's self-sufficiency in blood stocks, and
outlines the factors underlying the increased risk of hepatitis faced by
haemophiliacs.[1]
The timeline in Table 2.1 outlines the major events in the identification of
hepatitis C and the development of tests to detect the virus in blood. The events
listed are expanded upon in the remainder of the chapter.
Table 2.1: Timeline of history relating to
hepatitis C[2]
|
Australia
|
Date
|
International
|
|
|
1942
|
'Serum hepatitis' noted
in Second World War
|
|
|
1947
|
Two types of hepatitis
described
|
|
|
1965
|
Discovery of hepatitis
B surface antigen
|
|
Red Cross starts
screening for HBV
|
July
1971
|
|
|
|
1973
|
Hepatitis A virus
discovered
|
|
|
1975
|
Non-A, non-B hepatitis
described
|
|
Start of first
Australian post-transfusion study (published in 1982)
|
1979
|
|
|
|
April 1981
|
US Transfusion
Transmitted Viruses (TTV) study predicts ALT testing would reduce the
incidence of post-transfusion NANBH
|
|
|
August 1981
|
US National Institutes
of Health study predicts that ALT testing would reduce the incidence of NANBH
|
|
|
Nov 1981
|
Canadian Red Cross
Blood Transfusion Service advisory committee decides that ALT testing should
not be implemented as surrogate testing for NANBH
|
|
Post-transfusion study
of cardiac patients by Prof Cossart establishes risk of NANBH through blood supply at
1.7%
|
Jan 1982
|
|
|
|
March 1983
|
ALT screening
considered by US FDA, but no recommendation made.
|
|
|
1983
|
Committee of the
American Association of Blood Banks rejects implementation of ALT testing.
Even so, some blood banks introduce testing.
|
|
Red Cross adds
questions concerning high-risk sexual and injection behaviour to donor
screening
|
1984
|
|
|
First case of
transfusion related AIDS; introduction of uniform donor declaration by Red
Cross
|
July 1984
|
|
|
Surrogate testing using
anti-HBc for AIDS commenced in NSW
|
Oct 1984
|
|
|
Heat-treated Factor
VIII developed by Australian Red Cross
|
Nov 1984
|
|
|
|
Dec 1984
|
US TTV study predicts that anti-HBc testing would
reduce incidence of post-transfusion NANBH
|
|
|
1985
|
Introduction of HIV Ab
testing
|
|
Introduction of HIV
testing of donated blood
|
May 1985
|
|
|
|
July 1985
|
Preliminary data from
the Toronto incidence study show the incidence on NANBH to be
7.6 per cent
|
|
|
Nov 1985
|
Majority of US
fractionators begin to use ALT-tested plasma to manufacture blood products
|
|
|
Feb 1986
|
US FDA Blood Products Advisory Committee recommends
that all blood donations for transfusion be tested for both ALT and anti-HBc
as surrogate tests for NANBH
|
|
|
March 1986
|
American Association of
Blood Banks and American Red Cross issue a joint statement recommending that
blood collection agencies implement surrogate testing
|
|
|
April 1986
|
American Association of
Blood Banks board of directors decide that both ALT and anti-HBc testing of
blood donations should be implemented.
Report of results from
National Institutes of Health study predicting that anti-HBc would reduce
incidence of post-transfusion NANBH
Canadian Red Cross
Blood Transfusion Service advisory committee recommends against surrogate
testing for NANBH, pending further study of data from Toronto
incidence study and of the efficacy of HIV-antibody testing as a surrogate
test for NANBH
|
|
|
Nov 1986
|
Target date for
introduction of dual ALT and anti-HBc testing in majority of US blood banks,
even though testing not required by FDA.
|
|
Start of second
post-transfusion hepatitis study (published in 1995);
National Blood
Transfusion Committee does not support routine surrogate testing
|
1987
|
|
|
Queensland Blood
Transfusion Service begins surrogate testing
|
July 1987
|
|
|
Report on ALT surrogate
testing published in Queensland, Pathology
|
1988
|
|
|
|
May 1988
|
Identification of HCV
announced
|
|
BTS Executive
Subcommittee agreed to start testing for HCV antibody as soon as practicable
|
Dec 1989
|
|
|
Hepatitis C becomes
notifiable infection in States and Territories
|
1990
|
Screening test for
hepatitis C licensed in US
|
|
Super heat treated
Factor VIII available
|
Jan 1990
|
|
|
All transfusion
services had commenced screening for anti-HCV
|
Feb 1990
|
|
|
Agreement between CSL
and NBTC not to use anti-HCV repeat reactive plasma in the manufacture of
plasma products
|
June 1990
|
|
|
|
March 1991
|
US FDA requires
anti-HBc testing of blood donations to identify units contaminated with HBV
|
|
Second generation kit
introduced
|
May 1991
|
|
|
|
1992
|
Canadian Red Cross
implements second generation HCV antibody testing throughout Canada
|
|
NSW BTS reported that
only 30.8 per cent of donations found repeat reactive on anti-HCV screening
were positive on confirmatory testing
|
August 1992
|
|
|
Super heat treated
Prothrombinex becomes available
|
1993
|
|
|
Report on risk of post-tranfusion/
operative NANBH in Australia immediately before introduction of screening;
concluded 1st generation anti-HCV test detected about 85 per cent
of infective donations; and surrogate testing offered no additional advantage
Medical Journal of Australia
|
July 1995
|
|
|
Australian Red Cross
Blood Service established
|
1996
|
|
|
|
Nov 1997
|
Krever Commission
report released in Canada
|
|
Regulation of fresh
blood products commenced under the Therapeutic Goods Act 1989
|
2000
|
|
|
Introduction of Nucleic
Acid Testing for HCV
|
June 2000
|
|
|
National Blood
Authority established
|
2003
|
|
History and nature of Hepatitis C
2.2
'Hepatitis' means inflammation of the liver. It can result from overuse
of alcohol, reaction to certain medications or infection by bacteria or
viruses. There are several different viruses that cause hepatitis, such as
hepatitis A (HAV), hepatitis B (HBV) or hepatitis C (HCV). Each of these
viruses may produce similar symptoms and they can all infect and inflame the
liver. The main difference between the viruses is the mode of transmission, the
way they cause liver damage and the effect each has on a person's health.[3]
2.3
Hepatitis C infection can be either acute, characterised by a short-lasting illness, or
chronic, where hepatitis is
present for six months or more. Those with acute HCV are commonly
asymptomatic and may experience a mild flu-like illness. Some people, between
15 and 45 per cent (the higher proportion being in children), will clear
themselves of the virus within four to six weeks of infection. In the
remainder, chronic HCV infection occurs and causes the liver disease, chronic
hepatitis C. Most people with chronic HCV show few, if any, outwardly visible
symptoms. For this reason, many do not know they are infected. The symptoms
that may be evident are often general, and include fatigue, lethargy, nausea
and abdominal discomfort. The degree to which these symptoms may occur can vary
significantly.
2.4
During the acute phase, levels of the virus in the blood rise
dramatically until the body's immune response starts producing antibodies in an
attempt to destroy the virus. In many cases, the virus successfully tricks the
body into producing a poor antibody response. The infection is not brought
under control properly by the body and the infection becomes chronic.
2.5
The importance of HCV
infection lies in its persistence (or chronicity) and the liver disease it
causes. Once a person is chronically infected, the virus is almost never
cleared without treatment. In rare
cases, HCV infection can even cause liver failure. However, most instances of
acute infection are clinically undetectable.
2.6
The natural history of chronic HCV infection can vary dramatically
between individuals. Some will have clinically insignificant or minimal liver
disease and never develop complications. Others will have clinically apparent,
chronic hepatitis. Cirrhosis may develop in about 20 per cent of individuals
with HCV. This generally occurs at least 20 years after infection. Some
patients with cirrhosis will develop end-stage liver disease. A proportion of individuals
with cirrhosis resulting from HCV will also develop hepatocellular carcinoma
(primary liver cancer).
2.7
For patients with chronic HCV, it is difficult to predict who will have
a relatively benign course and who will go on to develop cirrhosis or cancer. Factors
promoting progression of HCV-related chronic liver disease include viral
genotype, age and sex of the person infected, alcohol abuse and whether the person is co-infected with
another virus.[4]
Certain findings on liver biopsy can help in predicting the course of the
disease.
2.8
The Barraclough Report noted that, based on studies of HCV infection
acquired through routes other than the receipt of contaminated blood or blood
products, it has been estimated that of all people with HCV antibodies, around
8 per cent would develop cirrhosis after 20 years following exposure, and 20
per cent would do so after 40 years. Rates of progression to liver cancer were
more uncertain, but were about 10 per cent of the rate of progression to cirrhosis.
Rates of progression to cirrhosis in people infected with HCV from a blood transfusion
are also generally much higher, as are rates of progression to cirrhosis in people
with established chronic liver disease.[5]
Progress of the disease is also discussed in Chapter three.
2.9
The public health impact of hepatitis C infection is substantial and the
socioeconomic costs to the Australian community are high. HCV also exacts a
high personal cost on sufferers as it has a long term impact on quality of life.
Further information on living with HCV is contained in Chapter five.
Hepatitis C epidemiology[6]
2.10
Hepatitis C is the most frequently reported notifiable infection in Australia.
It is estimated to affect about one per cent of the population, or 150,000 to
200,000 Australians, with an estimated incidence of 8,000 to 10,000 new
infections occurring each year. This compares to HIV with an estimated
prevalence[7]
of 15,900 cases and an incidence of 600 new cases per year.
2.11
The reported number of diagnoses of HCV infection has declined from a
peak of 20,465 in 2000 to 15,953 cases in 2002. The reported number of diagnoses
of newly acquired infection has declined from 672 cases in 2001 to 434 cases in
2002.
2.12
An estimated 225,000 people were living with hepatitis C infection in Australia
in 2002. This includes 133,000 with chronic HCV and early liver disease (stage
0/1), 29,000 with chronic infection and moderate liver disease (stage 2/3) and
6,900 living with HCV-related cirrhosis. An estimated 57,000 had hepatitis C
antibodies without chronic infection.
2.13
However, it is likely that many people with hepatitis C remain
undiagnosed. It is estimated that 210,000 people in Australia have been exposed
to the hepatitis C virus, of whom approximately 90,000 people live in NSW. Approximately
40 per cent of people in NSW who have been exposed to HCV are unaware of their
status.
2.14
The main mode of transmission of hepatitis C in Australia is through
unsafe drug injecting practices, in particular, the sharing and re-using of
injecting equipment. Approximately 80 per cent of infections are attributed to
the behaviour associated with injecting drug use, another 5–10 per cent to the
transfusion of blood products (prior to 1990) and the remainder to other forms
of blood-to-blood contact, such as non-sterile tattooing or other skin-incision
procedures.
2.15
Since 1990, all blood has been screened for hepatitis C and the risk of
transmission through the transfusion of blood or blood products in Australia is
now very low. The ARCBS modelling estimates the risk of contracting
post-transfusion HCV in Australia in 2000-2002 was 1 in 3,112,000.[8]
There is currently no vaccine against hepatitis C.
Number of people infected through blood transfusion
2.16
The Department of Health and Ageing (DoHA) stated that it is not possible
to obtain comprehensive or definitive figures on the number of people infected
with hepatitis C through blood transfusion. Many people with HCV are
asymptomatic and may therefore never have been diagnosed.
2.17
DoHA went on to state that 'it is accepted that a history of receiving
blood products before the beginning of blood-donor screening is likely to
account for a substantial proportion of HCV-infected individuals who are not
injecting drug users'. People with haemophilia who received fractionated plasma
derivatives before heat treatment procedures were implemented were particularly
at risk of being infected with HCV.[9]
2.18
The ARCBS provided the Committee with estimates of those living with
hepatitis C gained through blood transfusions. The ARBCS estimated that between
3,500 and about 8,000 Australians live with HCV infection derived through blood
transfusion, including an estimated 1,350 haemophiliacs.[10]
However, there is no formal reporting mechanism of post-transfusion hepatitis
in Australia, as pointed out by the ARCBS:
Australia does not operate a register where all suspected cases
of post-transfusion hepatitis might be found. Some countries have established
haemovigilance systems, which collect data in a central agency on all adverse
outcomes (infectious and non-infectious) from transfusion, investigate and
determine the cause...[I]n the early 1990s, all State and Territory governments
established hepatitis C as a notifiable disease...however, these local health
authorities do not necessarily record or confirm the route of transmission.[11]
The discovery of HCV
2.19
The transmission of blood-borne infections had been identified as an
issue with transfusions since their inception. With the development of methods
to monitor liver function, the term 'hepatitis' or inflammation of the liver
came into use. With the use of human transmission experiments and more advanced
knowledge of the disease, ‘infectious hepatitis’, which spread from person to
person by the faecal–oral route, and ‘serum hepatitis’, which was transmissible
by blood and blood products, were identified. In the 1970s infectious hepatitis
became known as hepatitis A and serum hepatitis as hepatitis B. Hepatitis B was
thought to cause post-transfusion hepatitis.
2.20
With the discovery of a protein called the B surface antigen (HBsAg),
scientists were able to find an antibody which reacted with this particular
protein. The antibody was subsequently used in developing tests to screen blood
donors for HBV. In Australia, a surface antigen test was developed in 1970 in
NSW and used throughout the country to screen donors. Professor Cossart noted
that routine screening greatly reduced the incidence of post-transfusion
jaundice globally. The ARCBS stated that, following the introduction of
screening, the post-transfusion rate of hepatitis declined by around 20 per
cent in the United States.[12]
2.21
The hepatitis A virus was identified in the faeces of a person with
‘infectious hepatitis’ in the early 1970s and HAV antibodies characterised in
1973. A test for antibodies (anti-HAV) then became available to study cases of
post-transfusion hepatitis that were negative for HBsAg.
2.22
However, while the incidence of post-transfusion hepatitis was reduced, screening
for both HAV and HBV failed to abolish the problem. People were identified with
sub-clinical post-transfusion hepatitis. This had a different clinical picture
from hepatitis A or B. In 1975 the name 'non-A, non-B hepatitis' (NANBH) was
coined. This term was used rather than hepatitis C because at the time it was
thought that more than one infectious agent was involved.[13]
2.23
In 1978, NANBH was successfully transmitted to chimpanzees. However,
many different groups failed to find a specific virus or a laboratory marker of
infection despite much intensive study. It was not until 1988 that a group of
scientists at the Chiron Corporation in the United States announced the
identification of the virus responsible for NANBH. A lay report appeared in Nature
and the scientific findings were published the next year.[14]
This was the first virus identified by the novel approach of gene cloning, and
the researchers named it ‘hepatitis C’.
2.24
Retesting of stored samples from past studies of post-transfusion
hepatitis soon showed that donors with antibody to the new agent had often been
implicated in transmission of non-A, non-B hepatitis. It is clear that HCV has
been the cause of liver disease for many decades (it has subsequently been
found in stored blood from 1948). It was therefore a newly recognised cause of
disease rather than a new virus.[15]
Hepatitis C in the blood supply
2.25
As stated above, it was noted in the 1970s that there was another agent
or agents that resulted in post-transfusion hepatitis. With the introduction of
testing for HAV and HBV, infection rates dropped but some recipients still
acquired hepatitis. In 1978 it was observed that, since the introduction of HBV
screening in the United States for donor blood, more than 93 per cent of cases
of post-transfusion hepatitis were attributable to NANBH.[16]
2.26
Several large scale studies were undertaken to ascertain the likelihood
of acquiring NANBH from blood transfusions under a defined set of
circumstances. Professor Cossart noted that there were wide discrepancies in
studies of post-transfusion NANBH in different countries. An Australian study
of cardiac surgery patients in 1982 returned one of the lowest rates while high
rates were observed in the United States, parts of Europe and Japan.[17]
2.27
In the United States there were great variations between blood
collection centres and studies in the early 1980s attributed this to the use of
blood derived from paid donors. Centres which used only volunteer blood had a
much lower rate of post-transfusion hepatitis than did those that relied
partially or fully on paid donors.[18]
2.28
The ARCBS also described two studies which were designed to define the
incidence of post-transfusion hepatitis in the United States and evaluate what
factors influenced its occurrence. The first, a multi-centre study published by
the Transfusion Transmitted Viruses (TTV) Study Group in 1981, showed an association between NANBH and
a heightened level of Alanine Aminotransferase, or ALT, an enzyme specific to
liver cells produced in response to hepatitis. An independent study at the
National Institutes of Health (NIH), also in 1981, confirmed the findings. In a
further series of studies there was an association between NANBH and the
presence of HBV core antibodies or 'anti-core', indicating prior HBV infection.
This issue was extensively reviewed in the Krever Report. The ARCBS stated that
there were predictions made, in the United States, that removing donors with
higher levels of ALT and positive for anti-core might reduce the development of
NANBH, by about a third, in recipients.[19]
Studies relating to surrogate testing are further discussed later in the chapter.
2.29
It was also known that there was a greater risk of transmission of NANBH
to haemophiliac patients because the risk of infection was compounded by the
use of pooled donations for the production of fractionated products. Witnesses
noted that, as a result, hepatitis was common in patients with haemophilia.[20]
(The use of fractionated products by haemophiliacs is discussed later in this
chapter.) However, it was generally considered that risk was acceptable because
there were such significant benefits in using Factor VIII and Factor IX
concentrates for the management of haemophilia.[21]
2.30
Following the Second World War, there was awareness in Australia, and
around the world, of the risk of hepatitis following transfusion. The ARCBS
stated that from the early 1970s the blood transfusion service consistently
warned doctors and hospitals of the risk.[22]
Studies into the transmission of NANBH were undertaken by Professor Cossart in
the early 1980s and by Ismay in the 1990s.[23]
Scientific meetings were also held in Australia which addressed NANBH.[24]
2.31
In the 1970s NANBH was considered to be a relatively minor disease with
the majority of patients being asymptomatic and without any sign of severe
impairment of liver function.
Background to blood and blood products
2.32
Blood is a major body tissue comprising plasma, a yellow,
protein-rich fluid that suspends formed elements: blood cells, white blood
cells and platelets. Plasma accounts for more than half of the total volume of
blood. It is around 90 per cent water and contains a very complex and not fully
understood mixture of proteins that perform many
bodily functions.
2.33
Organised blood transfusions first emerged in the 1920s, and only whole
blood was used. Over time, fractionation processes developed to the point
where, today, whole blood is rarely transfused. Fresh blood products are
perishable, with a shelf life of between 5 days (platelets) and 35-42 days (red
cells). Red cells are the most widely used blood product.
Table 2.2: Major fresh blood
components
|
Product
|
Main Uses
|
|
Red
cells
|
Replacement
of blood loss in trauma and surgery, and occasional treatment of anaemia.
|
|
Platelets
|
Control
of bleeding related to platelet deficiencies caused by disease (eg leukaemia)
or following severe haemorrhage or as a result of treatment of an underlying
malignant disorder
|
|
Cryoprecipitate
|
Treatment
of clotting factor and fibrinogen deficiency
|
|
White cells
|
Treatment of sepsis,
regeneration of blood cells after chemotherapy.
|
Source: Stephen Review, p.9.
2.34
Plasma products have a shelf life of between one and three years, and
can be divided into three main proteins; Albumin, Immunoglobulins, and clotting
factors.
Table 2.3: Principal plasma
products
|
Product
|
Main uses
|
|
Albumin
|
Treatment
of shock, burns, liver disease and kidney disease.
|
|
Immunoglobulin
for intramuscular injection
|
Temporary protection from infectious diseases such
as measles, rubella, and HAV.
|
|
Immunoglobulin
for intravenous injection
|
Replacement
therapy for primary immune deficiency disorders, such as
Guillain-Barre, and Kawasaki disease.
|
|
Immunoglobulin preparations with high levels of
specific antibody (hyperimmunes)
|
Treatment of tetanus or prevention of HBV, chicken
pox, haemolytic disease, the newborn or cytomegalovirus.
|
|
Factor
VIII concentrate
|
Haemophilia
A.
|
|
Other clotting factors
|
Other bleeding disorders such as Haemophilia B.
|
Source: Stephen Review, p.9.
Blood plasma and safety
2.35
The Barraclough Report provides an overview of issues concerning blood
plasma and safety. There are two types of plasma. Recovered plasma is obtained as
a by-product of whole blood collection and source plasma is obtained by
collecting whole blood from a donor, separating the plasma and returning the
cellular material to the donor. The standards under which recovered plasma is
collected are different from those that apply to the collection of source
plasma. In particular, the safety issues are influenced by the fact that recovered
plasma has to be subject to the same standards as plasma intended for direct transfusion.
Source plasma is subject to safety standards that are ultimately related to the
safety of the derivatives for which it serves as a raw material.
2.36
The principles underlying current concepts of the safety of
blood-derived therapeutics from infection by disease producing organisms, or
pathogens, are:
- the selection of donors from populations at low risk of carrying
transfusion-transmitted pathogens;
- the screening of such donors using appropriate laboratory tests;
and
- the treatment of the products using measures that eliminate any
residual pathogens.
Although desirable, it may not be
possible to have all of these principles in place concurrently.
2.37
Safety profiles differ for the two broad categories of blood-derived
therapeutics – plasma derivatives and blood components. Plasma derivatives are
produced from large donor pools. There is thus a greater likelihood of contamination
by blood-borne pathogens than for single donor products. However, plasma
derivatives are produced by industrial-scale manufacture and subject to
intensive processing and quality control. In the production process, steps to
eliminate pathogens can be instituted.
2.38
Viruses are the most important contaminants of plasma pools for
fractionation. The amount of viral contamination in a plasma pool depends on
several factors, and can be minimised through careful donor selection and
laboratory screening tests. Laboratory testing measures viral genomic material,
as well as the evidence of infection through, for example, antibody tests. Thus
the viral load for the important blood-borne pathogens such as HBV and HCV can
be reduced to very low levels.
2.39
Since the mid-1980s manufacturers have used various elimination steps
that eradicate the important viruses in plasma pools. Because of the large pool
size from which these products are derived, the mainstay of their safety from
viral infection is the ability of the manufacturing process to eliminate
viruses through deliberate steps and/or the biological features of the product.
2.40
Blood components, as
opposed to plasma derivatives, are usually derived 'under conditions in which
it is not possible to eliminate pathogens'.[25]
For these products, the main safety techniques are donor selection and
laboratory screening. The number of patients exposed to each product is much
smaller than for plasma derivatives, which assists their safety profile.
2.41
The Barraclough Report concluded that while the safety differential
between plasma derivatives and components has changed over the past twenty
years, the advent of viral elimination techniques have given plasma derivatives,
previously a higher-risk class of products than components, a superior safety
profile. This has been achieved with the identification of agents known to
cause disease, with the development of tests to identify these agents and with
the refinement of existing tests to enhance sensitivity.[26]
Surrogate testing
2.42
Surrogate testing, in the context of blood safety, refers to tests used
to detect viruses for which no specific test exists and to supplement specific
tests that are insufficiently sensitive.[27]
2.43
During the 1980s two surrogate tests for NANBH were proposed: testing
for abnormality of liver function through measurements of the level of alanine
aminotransferase (ALT); and testing for markers of previous hepatitis B
infection, the test for which was called anti-HBc. Professor Cossart noted that
the first test assumed that donors who were infective would have abnormal liver
function tests, while the second assumed that past exposure to one blood-borne
virus might predict a high probability of exposure to others.[28]
2.44
Witnesses reported to the Committee that before a specific test for HCV
was developed there was much debate as to the usefulness of surrogate testing.[29]
The Royal College of Pathologists of Australia stated for example, that the
decisions around surrogate testing were difficult and controversial as it is
neither sensitive or specific.[30]
The Australian Centre for Hepatitis Virology (ACHV) concluded that:
Consequently, any decisions made to introduce (or not) surrogate
screening tests were often based on interpretation of what information was
available, by individuals (blood bankers) who had the unenviable task of trying
to screen the blood supply for an unknown agent with no tools.[31]
Arguments for surrogate testing
2.45
A number of witnesses submitted that surrogate testing should reasonably
have been introduced across Australia from around 1986. It was argued that this
form of testing represented a useful indicator of HCV status, and that its
introduction would have prevented at least some infections through transfusion.[32]
It was also noted that surrogate testing was introduced in some other
countries, and in Queensland in 1987.
2.46
Those supporting the introduction of surrogate testing pointed to
studies conducted in the United States which were reported in 1981. The Transfusion
Transmitted Viruses Study reported an association between elevated ALT in
donors and the development of NANBH in blood recipients. The study predicted
that by excluding donors with elevated ALT, 40 per cent of NANBH might be
prevented at a loss of 3 per cent of the donor population. This low degree of
supply loss was another advantage of using ALT as opposed to anti-HBc. The
investigators concluded that a 'compelling argument' existed for ALT screening
and exclusion to take place.[33]
In his submission to the Inquiry, Professor James Mosley, the Project
Coordinator of the TTV Study, recalled reporting his findings at a conference
in Brisbane in 1978. Professor Mosley reported that a number of blood bankers,
including at least one senior Australian Red Cross employee, were in
attendance.[34]
2.47
A study by the National Institutes of Health in 1981 found an almost
identical outcome predicting donor exclusion based on elevated ALT might
prevent 29 per cent of transfusion associated hepatitis at the loss of approximately
1.5 per cent of the donor population. However, this study also noted the high
incidence of false negative and false positive results, and did not recommend
the introduction of ALT testing. It was stated that:
The ALT testing of donors is thus a tenuous balance between risk
and benefit. The balance shifts toward testing when one considers that
approximately 30 per cent of [post-transfusion hepatitis] might be
prevented...but this is tempered by the realization that 70 per cent will not be
prevented and that the prevention of 30 per cent is in some doubt unless
confirmed by a randomized clinical trial. The balance also shifts away from
testing when one considers the estimated additional $20 million in the annual
cost of blood to the United States alone and the potential national loss of
45,000 donors and more than 90,000 units of blood. It is a difficult equation,
whose solution will require thought and planning.[35]
2.48
However, the NIH findings in relation to anti-HBc differed to those for
ALT. The NIH report concluded:
If, as predicted, surrogate screening of blood donors could
prevent approximately one third of these cases, then this could represent an
annual reduction of 50,000 cases of hepatitis and 2,500 cases of cirrhosis. The
potential to achieve this degree of disease prevention now appears to outweigh
the disadvantages inherent in the adoption of surrogate tests for the non-A,
non-B virus carrier state.[36]
2.49
Later the TTV and NIH studies were re-analysed and an association was
shown between the anti-HBc marker in donors and the development of NANBH in
recipients.[37]
2.50
The Queensland Government was unable to provide the Committee with
information about the decision to introduce surrogate testing. However, Dr Catherine
Hyland, of the Blood Transfusion Service in Brisbane, published a study in
1988 which concluded, inter alia:
The recent judgement in a legal suit that concerned the
Queensland Red Cross Blood Transfusion Service has indicated that, provided the
transfusion service is implementing screening procedures appropriate to
published professional knowledge at the time of transfusion, there should not
be a case for negligence at law...[I]n the light of this experience, and given
the development of an assay that is cheap and convenient, it was decided that
concern regarding chronic effects of NANB hepatitis outweighed the arguments
against implementation of surrogate testing.[38]
2.51
The Haemophilia Foundation Australia (HFA) commented that, 'it appears
that issues such as test sensitivity and specificity, cost and fears about
reduced blood supply were considered more important than the seriousness of
hepatitis'. The HFA went on to argue that 'if any kind of testing was available
that could have potentially saved people from a life threatening virus, efforts
should have been taken to implement these. Decisions based on cost
effectiveness do not stand the test of time'.[39]
Arguments against surrogate testing
2.52
A number of arguments were put to the Committee as to why surrogate
testing was not supported. First, it was argued that surrogate tests are no
substitute for specific tests such as antibody tests. Because of the lack of
sensitivity and specificity, it is difficult to ascertain their effectiveness
in identifying the blood donations that should be excluded.[40]
2.53
In relation to the two surrogate tests proposed for NANBH it was pointed
out that there were problems with both tests. For ALT, it was argued that, by
its nature, it was not specific to NANBH. There were a number of reasons why
ALT levels may be raised, including individual lifestyle factors such as
exercise, alcohol, use of many common medications and simple obesity.[41]
The Barraclough Report noted that:
ALT measures a normal liver enzyme. This is not a measure of the
presence of a particular hepatitis virus. Rather, elevated ALT levels may be a
sign of liver inflammation, commonly caused by hepatitis. However, as ALT
levels are affected by many drugs, including even modest amounts of alcohol,
many units of non-infective blood gave abnormal results. Furthermore, at least some
infective units had normal values. In addition, an ALT elevation may not mean
the person has any medical abnormality.[42]
As a result there would be high levels of donors rejected
unnecessarily.
2.54
There was also considerable debate at the time about the significance of
raised ALT levels and the ALT cut off level where blood should be discarded.
For example, it was known that ALT levels could vary even where the individual
was a carrier of the NANBH agent. The person could thus have an ALT level above
the cut off on one day and a lower ALT level on another day.[43]
Professor Geoff McCaughan, in his submission to the Committee, pointed to a
number of reviews published in the mid 1980s which addressed the inadequacies
of surrogate testing.[44]
2.55
Professor Cossart referred to a review of the issue of surrogate testing
over the past three decades published in 2000 that concluded that 'despite its
conceptual appeal, ALT screening had never been substantiated as a routine
measure to prevent post-transfusion NANB hepatitis, and its introduction was
driven by concern about the emerging problems in recipients rather than
evidence of its efficacy'.[45]
2.56
In evidence from CSL, Dr Darryl Maher provided the Committee with a
graph generated by the Therapeutic Goods Administration. The graph, reproduced
as Figure 2.1, plots the course of viral load in an individual over the days
following infection. Dr Maher's explanation of the graph and its consequences
for ALT testing is worth quoting at length:
This is from time zero, the point at which the individual is
infected, and this is the course of the infection in days, out to 100 days. The
Y axis is the level of virus in the blood. That axis is actually on a
logarithmic scale, which means that at each point going up the Y axis we are
talking about tenfold more viruses. At this point down here there may be, say,
100 viruses per millilitre; up here, it would be of the order of 10 million
viruses per millilitre – so many, many thousandfold more. After infection,
within about a 10- or 11-day period, the virus starts to appear in the
bloodstream in the individual – and this is it going up here. The tests that
can detect that are the NAT tests, which you have heard about, because they are
measuring the virus itself.
With regard to the earlier tests, let us start with the
surrogate testing, the ALT marker. That is a marker of inflammation in the
liver, so it only goes up once the infection has taken hold and the liver has
become inflamed. You can see the ALT peak on this graph here which shows that
it is some 50-odd days after the infection before the ALT starts to go up. So
for donors who may have been infected and are at risk of transmitting you have
this 55-day period with extremely high titres of virus, and none of these
tests—the ALT or, for that matter, the antibody tests – are able to detect it.
The unfortunate irony, in a way, is that the time when the
antibody takes off and the ALT is coming up is the time when the level of virus
actually starts to fall. So the level of virus in the group that are positive
for ALT is about 10,000-fold less than the level of virus in this group of individuals
who are in the incubation period before their test becomes abnormal. We are
talking about 10,000 to one, so if you have got a 10,000-donor pool you only
need to have one person in this period for there to be as many viruses as
having all 10,000 of them with a positive ALT test.
That is how dramatic the difference is in the level of virus
during that course. This information is in retrospect and it was not available
to the committee making decision at the time. I think other reasons drove the
decision back then. What I am saying is that, in retrospect, it is very clear
that ALT testing would not have reduced the risk of transmission by these
concentrates.[46]
Figure 2.1
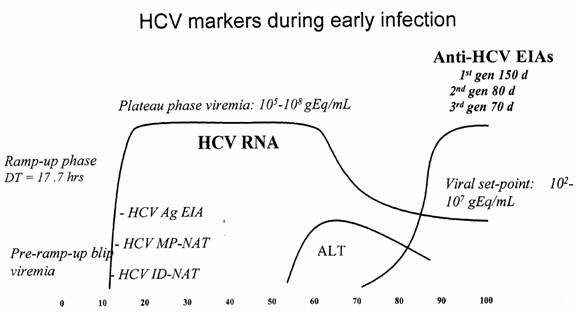
Source:
TGA additional information, tabled by CSL on 5.4.04.
2.57
In relation to anti-core testing, Professor Cooksley noted that it had
the advantage of being positive or negative rather than being a continuous
variable. However, the disadvantage was the high rate of false positive and
false negative results. Anybody with a past exposure to HBV would be
automatically excluded. Thus people from the Mediterranean countries, Eastern Europe,
the Middle East, Asia, Pacific region, Africa and South America would have a
high likelihood of being excluded as HBV is common in those regions. However,
only about half of the HCV-positive donors would be excluded, since the test
relies on previous exposure to HBV.[47]
2.58
The need for surrogate testing was also questioned as the studies
supporting the introduction of testing were derived from the United States,
where the epidemiological context differed significantly from that of Australia.[48]
This raised the question as to whether it was appropriate or necessary to
introduce surrogate screening in Australia. The Barraclough Report stated:
The greatest potential benefit from using surrogate tests was in
countries where the risk of transfusion transmitted hepatitis was highest,
notably in countries that used blood and blood products from paid donors.[49]
2.59
Professor McCaughan also pointed out that not only did Australia have a
volunteer donor system but also a successful HIV screening questionnaire
programme had been introduced in Australia while in the United States neither
precaution was taken.[50]
The Barraclough Report also commented on the significance of HIV questionnaires
and found that:
The majority of data supporting the efficacy of surrogate
testing were obtained before the introduction of donor screening by
questionnaire and serological testing for HIV. Both of these activities were
likely to have significantly reduced the effectiveness of the surrogate
screening protocol by excluding a significant proportion of the same risk
group.[51]
2.60
The ARCBS submitted that 'Australian blood bankers took all questions of
safety extremely seriously and thoroughly reviewed and considered the
"surrogate marker debate" as it evolved in the United States, Europe
and the United Kingdom'. However, it was decided, through the National Blood
Transfusion Committee, not to recommend the introduction of surrogate testing
'following an evaluation of the scientific evidence for surrogate testing
because the evidence that it would be effective was not convincing'. Surrogate
tests were considered to be 'blunt and inaccurate tools with the potential to
create blood shortages without any demonstrated benefit to public safety'.
Further, surrogate tests had not been proven to be effective in reducing
post-transfusion hepatitis.[52]
2.61
In relation to the introduction of surrogate testing in Queensland, the
ARCBS stated 'the fact that the BTS in Queensland, having reviewed the same
international data and arguments as the other services, reached a different
conclusion from the remaining states is evidence of the highly controversial and
inconclusive nature of the "surrogate marker debate"'.[53]
Surrogate testing internationally
2.62
The inconsistent approach taken internationally was borne out by
evidence on the introduction of surrogate testing overseas which was provided
to the Committee. For example, in the United States in 1983 a report from the
American Association of Blood Banks concluded:
While we share the desire of the entire medical community to
reduce the incidence of transfusion associated hepatitis, we believe that
currently available evidence does not justify either universal testing of donor
blood for ALT or the rejection of donors who have elevated levels. Therefore,
at this time we do not advise routine donor testing for ALT as a means of
reducing the incidence of non-A, non-B hepatitis.[54]
2.63
However, the US Blood Banks adopted surrogate testing at various times
up to mid 1987. The US Food and Drug Administration blood products advisory
committee found that surrogate testing should be implemented. Despite the
recommendation of its own blood products advisory committee, and introduction
of surrogate testing by Blood Banks, the FDA did not issue a regulation
requiring anti-HBc testing of donated blood until 1 March 1991, and then for the purpose of identifying units contaminated with HBV, not HCV. The FDA never
issued a regulation requiring testing for ALT levels, and only a 'handful' of US
blood centres implemented it as a matter of course. However, the American
Association of Blood Banks recommended in 1986 that testing be introduced and
this occurred in 1986-87.[55]
2.64
Few other countries introduced surrogate testing in the mid 1980s. The United
Kingdom did not implement surrogate tests. The average rate of post-transfusion
hepatitis was believed to be less than one per cent, so low that British blood bankers
questioned whether it was cost effective to implement even anti-HCV testing, when
it became available.[56]
No European countries performed anti-core testing and only parts of Germany and
Italy conducted ALT testing. The ARCBS noted that Germany had introduced ALT
testing in the 1970s but it still had a very high rate of post-transfusion
hepatitis.[57]
2.65
In May 1987, the Council of Europe's Committee of Experts on Blood
Transfusion and Immunohaematology concluded that:
Arguments against the introduction of surrogate testing include
the variability of data from one country to another, the non-specific nature of
the tests proposed, loss of apparently healthy donors, difficulty in follow up
of the donors and the continuation of transfusion-transmitted NANBH in spite of
the tests.[58]
2.66
Those in support of surrogate testing argued that the prospect of a
reduction in the supply of blood (owing to the need to discard blood which may
nor may not have contained HCV) was a major factor in the decision not to
introduce surrogate testing.
2.67
The ARCBS stated that the level of donations was a 'major concern' as it
was estimated that at least five per cent of voluntary blood donations would be
rejected even though they were mostly expected not to be infectious. The false
positive result from the ALT test might occur if the donor was overweight, or used
alcohol heavily before donating, or was taking certain medicines. The ARCBS
also noted that it was during this time that there was concern about the
adequacy of the blood supply as the AIDS epidemic had led to a fall in
collections.
2.68
In addition, the Blood Transfusion Services were mindful of causing
needless alarm in donors by advising them that they may have contracted
hepatitis. Many donors would have been referred to medical practitioners for
investigation and possibly even a liver biopsy, a procedure with risks of its
own, even though the great majority of donors would be healthy.
2.69
The ARCBS also argued that such a move might also have been counterproductive,
as lost donors would need to be replaced and a consequent increase in new
donors would have brought an increased risk. New donors were known from experience
with HIV and HBV to have much higher rates of infectious disease markers than
repeat donors were.[59]
2.70
In Queensland, during the three year period of ALT testing over 4,400
donations were estimated to have been discarded. Many new donors were required
and the ARCBS stated that this created problems for the Queensland BTS. It
added that, in retrospect, it was clear that 92 per cent of the blood Queensland
rejected was in fact good blood. The ARCBS concluded that 'essentially
surrogate testing was casting a very wide net in which you may have caught just
a few of the infectious donors but also a lot of good safe donors got caught as
well'.[60]
2.71
It was also suggested in evidence that the costs associated with
surrogate testing bore an impact on decisions as to its use.[61]
The Tainted Blood Product Action Group (TBPAG) claimed that the ARCBS had:
[a] desire to place commercial considerations before the primary
responsibility of maintaining a safe blood supply...[62]
2.72
The Committee
received evidence from the ARCBS addressing the cost of surrogate testing as
follows:
We have examined
records from the relevant time held by ARCBS nationally and found only one
specific estimate. That was from NSW, the largest Blood Service. NSW estimated
that the cost of conducing ALT tests alone for the year 1987-1988 would have
been approximately $250,000. This figure did not include any costs associated
with replacing lost donors. Based on NSW representing about 33% of Australia's blood collection at the time, one
could therefore project the total Australian costs for ALT testing might have
been in the order of $750,000 - $800,000 per annum.[63]
With respect to
anti-core testing, the ARCBS went on to provide the following:
The core antibody
test was estimated by Queensland to cost more than ALT testing. In June
1992, it was referred to as having been costed in 1987 at $250,000 per annum
for Queensland. Based on Queensland representing approximately 17% of
Australian collections in the late 1980s this would equate to a cost of about
$1.47 million nationally per annum.[64]
2.73
The ARCBS strongly rejected the claims concerning costs, arguing that
cost issues were never a consideration by the (then) Australian Red Cross in
their assessment of the usefulness of surrogate testing in the Australian
context:
Commercial considerations played no part in the decision making.
It is important to note that cost was not a consideration and has never been
claimed to be an issue in the decision making on this surrogate testing in Australia.
Red Cross funding at that time was not reliant on the volume of collections
therefore any fall in collections did not affect funding.[65]
2.74
Appearing in Sydney, Professor Barraclough summarised what he considered
to be an extremely difficult decision making process:
My view is that the issues were considered effectively by quite
serious and concerned people who were trying to balance quite momentous
national issues in effect but without adequate scientific knowledge to give
them the certainty and security that they would normally have when taking
decisions of this nature...[T]he fact that Australia was so early in introducing
the first [antibody] test says that people were taking those issues of public
safety very seriously.[66]
2.75
Professor Burrell of ACHV concluded:
In looking back now to assess what might or might not have been
instituted at a certain point in time, two further considerations apply. (i)
Armed with our current knowledge about HCV, it is hard to fully appreciate the
uncertainty and lack of quantitative information available before 1989, and
also in the period 1989-1992. Furthermore, the number of false starts and blind
alleys that occurred during the 1980’s had created a certain sense of caution
against immediately adopting possible new measures. (ii) There have been
changes in society’s tolerance of risk from blood transfusion. Prior to the
1980’s, the measurable risk of hepatitis from blood transfusion was
acknowledged and enormous efforts were made to reduce this to a lower level,
compatible with the requirement to maintain blood supplies. The success of
these efforts, the reduction in the risk of transfusion-transmitted HIV, and
the institution of nucleic acid screening to even further reduce the
transmission of specified agents, have all contributed to a current climate
where, in balancing cost-benefit issues of blood safety versus possible blood
shortage, a particularly high expectation is now required for safety from
transfusion-transmission of hepatitis.[67]
2.76
Dr Baird expressed a general view of the majority of medical witnesses,
putting it this way:
...[I]nternationally there was some wide disparity over what was
and what was not appropriate. Some countries were performing testing; others
were not. It was purely on the evidence that some people evaluated different
evidence in different ways; it was not a universal approach internationally. In
retrospect it is easy to look back and say, 'Ah, how progressive' but on the
other hand it was not retrospect at the time.[68]
2.77
The Royal College of Pathologists of Australia stated that surrogate
testing may have decreased, though not eliminated, the transmission of NANBH
but 'this does not mean that the introduction of such testing was appropriate'.
The RCPA commented that factors in the decision would be:
- the predicted decrease in the transmission of hepatitis by the
introduction of surrogate testing;
- the percentage of donors deferred on the basis of surrogate
testing and the impact that this would have on the adequacy of the blood supply
- the impact on the deferred donors themselves, especially as many
would not actually have significant illness.[69]
The possible prevention of
hepatitis C infections by earlier implementation of surrogate testing and donor
deferral
2.78
Submissions from the ARCBS and the paper prepared by Professor Cossart
for the DoHA addressed the issue of the number of infections which may have
been prevented had surrogate testing and donor deferral been implemented
earlier.
2.79
The ARCBS stated that 'it is almost impossible, hypothetically, to
quantify the potential benefit of surrogate testing or the impact on the blood
supply of its introduction in Australia'. Rather the ARCBS provided evidence on
the countries that did introduce surrogate testing and their retrospective view
of the benefit.
2.80
In the United States various studies found that:
- 91 per cent of US donors with elevated ALT and 95 per cent with
anti-core were HCV negative;
- the introduction of surrogate testing in 1986-1990 resulted in
little difference in the proportion of multi-transfused patients who developed
HCV;
- the most significant drop in the incidence of NANBH occurred with
the exclusion of paid donors and the introduction of the HBV surface antigen
test in 1970; and
- the combined effect of ALT testing and implementation of anticore
as a surrogate test in 1987 was a drop in the incidence of NANBH from 5.5 per
cent in 1981 to 4.1 per cent. This
change in 'background risk' was significant.[70]
2.81
The ARCBS noted that reductions in post-transfusion NANBH occurred in
countries without the introduction of surrogate testing. For example, the rate
in Canada declined from 9.2 per cent the early eighties to 3.2 per cent in the
late eighties. Other studies from Australia and Europe showed similar results.
It was believed that reductions in the risk of NANBH were due to the
introduction of other preventative measures. The major measures were the
limiting of the amount of blood given to an individual; phasing out of paid
donors; and more intense screening of volunteer donors.[71]
2.82
Professor Cossart stated that some anti-HCV positive donations would
have been rejected and a proportion of post-transfusion NANBH cases prevented
had surrogate testing and donor deferral been implemented during the 1980s. The
number of cases prevented and overall effect would have depended on the actual
level of the cut off level used to define ALT abnormality; the ethnic and
social composition of the donor panel of the time, and the actual rate of post-transfusion
NANB hepatitis following transfusion of units retained or rejected.
2.83
Professor Cossart noted that it is not easy to make an assessment in
retrospect and even at the time as surrogate testing was only one of four major
strategies used during the 1980s to reduce the risk of NANBH after blood
transfusion. In addition, few large scale trials on the effect of each measure
were undertaken.
2.84
Professor Cossart estimated the hypothetical benefit in Australia from
exclusion of donors using surrogate markers:
If surrogate testing for both raised ALT (>50IU/L) and
anti-HBc alone had been introduced during the late 1980s approximately 512
(0.091%) units would have transmitted HCV each year compared with 615 (0.11%)
had the same number of donors been deferred on the basis of an arbitrary marker
such as the initial of their surname.
The number of cases of hepatitis C prevented would have been
substantially less as most patients receive multiple units of blood. Factors
which would have attenuated the impact are that the risk of persistent
post-transfusion HCV is less than 25 per cent of the risk of transmission and
the risk of chronic HCV related liver disease is still lower.[72]
First generation test for hepatitis C
2.85
The molecular characterisation of the hepatitis C virus in 1989 led to
the rapid development of a test for antibody to the virus. Epidemiological
studies quickly revealed that HCV was the cause for at least 80–90 per cent of NANBH.
The first generation antibody test was subsequently shown to be capable of
preventing the transmission of 75 per cent of transfusion-transmitted HCV, the
major source of non-A, non-B hepatitis.[73]
2.86
The first tests designed to measure anti-HCV antibodies became available
commercially in late 1989. The first HCV kits measured antibody to the C-100
antigen, which is not part of the infectious HCV particle itself, but is made
in infected cells as the virus grows. Antibody against the C-100 antigen
appears irregularly in acute infection but is usually present in chronic
carriers of HCV. Antibodies of this type do not protect against infection, and
may cross-react with antigens induced by other related viruses. Professor Burrell
stated:
The first screening test used a very small area of the antigens
of the virus and the technology was not as good at dealing with cross-reactions
or non-specific binding patients antibody. So some patients in whom the
antibodies that had developed did not happen to match up with the narrow range
of antigens in the test would have had true antibody but it would not have come
up in the test, and that would have given a false negative result. Then there
would be other patients in whom the screening test would give a positive
reaction. The reason would not be that they had the hep C antibody; the reason
would be that they had some other kind of reactivity, that the plasma was
sticky or some other unrelated reason.[74]
2.87
The Barraclough Report noted that for many months after the introduction
of the tests, there was no independent means of confirming a positive result
and this placed transfusion services worldwide in a difficult position. Initial
screening of donors revealed a higher rate of positive test results than would
be anticipated given the rate of clinical post-transfusion hepatitis. For
example, the ARCBS stated that, 'in the first phase, 70 per cent of the people
who reacted on the test were false positive; so they did not have HCV at all'.[75]
There was also very little knowledge about the significance of a positive test
result in terms of the risk of developing significant liver disease or of infectivity
to contacts in everyday life. There was consequently no consensus about the most
appropriate approach to counselling donors who tested positive for anti-HCV antibodies.[76]
2.88
Australian blood transfusion services decided to introduce screening of
donations using the first generation C-100 test in November 1989 with
commencement of use of the kits by all Blood Transfusion Services in Australia
by 19 February 1990. It was expected that confirmatory tests would rapidly
become available given the volume of research being conducted by the Chiron
group and others, particularly in Japan.
2.89
Australia was one of the first countries to use the first generation
test kits, with most countries introducing the kit during 1990-91.
Specifically, these included France and Finland as of May 1990, Canada in June
1990, the USA (Blood Sector) between May and November 1990, the United Kingdom
by September 1991 and Denmark by early 1991.[77]
2.90
While there were some reservations expressed on the accuracy of the
first generation test, Professor Burrell commented:
I do not have the percentages in front of me as to what we think
their performance was compared to the best standard now, but I am fairly sure
that even the first generation tests would have been well in the range of 75
per cent to 95 per cent reliable compared to what we have got now, which is
just an extraordinarily large improvement on anything that surrogate markers
were attempting to do. The introduction of the first generation test in 1990
was an absolute watershed, moving from being in the dark blindfolded to having
a fairly reliable window on what was going on.[78]
2.91
This test is estimated to have prevented 75 per cent of
blood-transmitted HCV in the USA, or 40,000 patients per year.
Testing and exclusion of products destined for
fractionation
2.92
It is clear that there was a significant divergence of scientific
opinion and debate internationally as to the use of plasma testing positive to
the newly developed anti-HCV test for the manufacture of plasma products, and
the relative safety of immunoglobulin produced with such plasma. Based on the
incomplete scientific knowledge of the time, and after wide consultation and
detailed discussion of the conflicting evidence, the decision was taken to
allow plasma that tested positive to the first generation anti-HCV test to be
sent to CSL. This occurred from February 1990, when anti-HCV testing was
introduced, through to July 1990.[79]
2.93
The Expert Advisory Group chaired by Professor Barraclough found that
positive plasma was allowed to be fractionated for the production of specific
products, none of which had been associated with hepatitis transmission
provided that particular manufacturing processes were followed. The Group also found
that plasma testing positive continued to be stored with CSL until July 1991
for use in research, but that the stockpile was destroyed by May 1994.
2.94
The decision to allow plasma which tested positive to be fractionated
for certain products was in accordance with the stated policy of the United
States Food and Drug Administration, which considered that the immediate use of
the first generation anti-HCV test to exclude plasma for further manufacture
was premature.
2.95
However, further consideration by the Red Cross in April and May 1990
led to a reversal of this decision. One key consideration was the publication
in The Lancet in May 1990 of a letter from the Director of the Scottish
Red Cross Blood Service, Dr John Cash, who considered that a continuation
of the FDA's policy of inclusion of plasma which tested positive could be
regarded as 'a major breach of good manufacturing process'.[80]
Testing and notification policy in the introductory
phase[81]
2.96
The Barraclough report commented that in 1990, first generation antibody
tests returned a large number of false positive results. Confirmatory tests for
hepatitis C were not available for many screened anti-HCV positive donors, particularly
in the first three quarters of 1990, and this created difficulties in identifying
true positive results. This also lead to greater difficulties in counselling
the donors who tested positive. As a result, the Blood Transfusion Service
Executive Sub-committee decided in a meeting on 22–23 February 1990 that donors
who were repeatedly reactive to anti-HCV screening would not be notified in the
first instance. It was agreed at that meeting that donors who were repeat reactive
to anti-HCV and had a raised (ALT) at a subsequent donation would be notified
and referred to a gastroenterologist.
2.97
As an interim measure, donations testing positive in the C-100 test were
retested by the same means. Units which tested positive a second time were
withdrawn from routine use and sample was stored for confirmatory tests in the
future. An additional test using an assay was called recombinant immunoblot assay
(RIBA) was available in limited quantity during the Phase 1 period. The RIBA
confirmatory testing commenced in NSW on 3 September 1990, as soon as the kits
were commercially available. A small number of trial kits had been provided
earlier in the year by Ortho Diagnostics for research purposes.
2.98
Donors whose blood repeatedly tested positive to hepatitis C screening
tests continued to donate for plasma fractionation products only, until July
1990. Donors were not deferred from making donation until tests that could
confirm their HCV status became available. These tests were not universally
available until towards the end of 1990, although the first tests became
available in September 1990.
2.99
Donor follow-up included further testing at three and six months,
including an interview with a blood transfusion service medical officer, to establish
if they were still infected.
2.100
The management of anti-HCV (positive) repeat reactive donors was
discussed again at a BTS Executive Sub-committee meeting on 18 July 1990. At the meeting it was noted that the majority of blood transfusion services were
abiding by the February decision of the BTS Executive Sub-committee. It was agreed
that donors should be referred to an appropriate clinician if they were
repeatedly reactive to HCV testing as well as showing raised ALT level, and
were positive to a confirmation test. It was acknowledged at this meeting that
confirmatory tests for HCV antibody were not always available. When confirmatory
tests became available and confirmation of HCV positive status was achieved,
such patients were counselled, referred to an appropriate clinician and
deferred from donation. From December 1990, following discussion at the BTS
Executive Sub-committee, repeatedly reactive screening tests were considered as
a basis for deferral until true confirmatory tests became available.
2.101
In evidence to the Committee, one witness related his experience of
blood donation, expressing concern at being encouraged to donate even after his
positive hepatitis status was confirmed.[82]
Indeed, the Barraclough Report indicated that, depending on the State or
Territory, antibody-positive plasma continued to be shipped to CSL as late at
July 1991. However, the Expert Advisory Group concluded that, while donations
may have been made, blood testing positive almost certainly was not used by CSL
to produce plasma products.[83]
2.102
In a supplementary submission to the Committee, the ARCBS reported that
a study was conducted during 1990 to investigate the efficacy of the first
generation HCV antibody test, and that some donations made after July 1990
which tested positive to the test were used in that study. The ARCBS indicated
that contributors to this study were advised that their donations may also be
used for fractionation into products carrying no risk of transmission post
manufacture.[84]
ARCBS also stated that any plasma testing positive after July 1990, not used
for the study, was stockpiled at CSL with a view to its use in the production
of a new hyper-immune anti-HCV immunoglobulin. This stockpile was subsequently
destroyed, the project unrealised.
Second generation testing
2.103
With advances in the understanding of the hepatitis C virus and refinements
in molecular technology, a second generation test based on a series of antigens
derived from other HCV genes was developed in 1991. Professor Burrell noted
that the new tests improved the range of antibodies they detected and could
detect closer to 100 per cent of true infections. Approximately half of the
donors who tested anti-HCV positive in the first generation test remained
positive in the second.
2.104
Professor Burrell went on to state:
Early on we did not really have any other yardstick.
Subsequently, what has become more and more available is a means to detect the
virus rather than the antibody. The presence of the antibody usually would be a
reflection that the patient had been infected. If infection invariably leads to
persistence, as it does with HIV, you can take the presence of antibody as proving
the patient is now infected. But, with hepatitis C, we believe that only 65 per
cent to 85 per cent of people with antibody are truly infected still and the
rest have their antibody but have cleared the virus.[85]
Testing for hepatitis C today
2.105
In testing for hepatitis C, a sample of blood is taken and tested to
determine whether the person’s body is producing antibodies to the virus. After
exposure to the virus it can take up to six months before antibodies can be
detected. This is known as the window period.
2.106
An HCV RNA test, sometimes called PCR (polymerase chain reaction test), is
now used. This tests for the presence or absence of the virus itself (the viral
RNA). The test is generally used when assessing people for treatment and can
also be used where an antibody test result is indeterminate. Professor Burrell
stated:
There are still problems with that test because that only has a
certain sensitivity and, if a patient has a fluctuating level of virus, there
may be times when the level goes under the sensitivity level and then comes up
again. So they may appear negative and then be positive a week later.[86]
2.107
As to the overall quality and accuracy of testing in 2004 by the ARCBS, Professor
Elizabeth Dax, Director of the National Serology Reference Laboratory, which
is charged with assuring the quality of HIV and HCV tests in Australia, stated:
Not only does the ARCBS strive to put in place the most
appropriate methods but they are certainly followed rigorously, in a
batch-by-batch way, on a continuous basis. I think all the tests and
innovations have been put in place not only promptly but in a very controlled
manner and in such a way that they have been able to be checked on a continuous
basis.[87]
Conclusions
2.108
The Committee received evidence that there was widespread controversy
surrounding the use of surrogate testing in respect of hepatitis C. The
Committee considers that this inhibited the ability of authorities around the
world in making decisions on its implementation. Australia was no exception,
and a good deal of time and resources were spent in search of a definitive
outcome, to little or no avail.
2.109
There is evidence to suggest that the relevant authorities in Australia
could have instigated surrogate testing prior to the introduction of the
antibody test in 1990. However, the Committee was presented with much
compelling evidence as to why surrogate testing was not introduced.[88]
It seems to the Committee that, based on the information available at the time,
it was open to the relevant bodies to take the decisions they did. It is in
this context that the concept of equipoise arises, whereby, to quote Professor McCaughan:
If on the balance of the evidence you do not know what to do,
then either choice is ethically acceptable.[89]
2.110
The difficulty
associated with the decision making process at the time was also acknowledged
by the Hepatitis C Council on New
South Wales:
On balance while we
regret, in the strongest possible terms, that hepatitis C infections arose as a
result of this decision, we do not believe that negligence or at fault
activities occurred.[90]
2.111
The Committee therefore considers that, at the relevant times, decisions
made in relation to surrogate and antibody testing were not inappropriate. The
Committee is confident that due consideration was given to pertinent evidence
at relevant times, and that decisions were reasonable in the circumstances.
Australia's self sufficiency in blood stocks
2.112
The Department of Health and Ageing (DoHA) stated that the aim of
national self-sufficiency in blood supply has been part of official Australian
policy since 1975.[91]
The policy for self-sufficiency arose out of an international concern that some
commercial fractionators were buying plasma from persons in developing
countries. This posed a risk both to the paid donors and to the recipients of
products made from plasma.
2.113
Australia's aims in relation to blood and blood products are set out in
the recent National Blood Agreement between the Commonwealth and
State/Territory Governments where one of the policy aims is 'to promote national
self-sufficiency'.[92]
2.114
The Committee heard that, in developed countries such as Australia, self
sufficiency could be taken to imply a sufficient supply of both fresh blood
components and fractionated plasma products such as albumin, clotting factors and
immunoglobulins. This would normally be achieved through a national blood
program without the need to source products from other countries. A blood
donation rate of 50 per 1000 population is the general minimum donation
rate required for a developed country to meet this objective. In Australia,
this translates to around 20,000 donations per week being needed to keep
supplies at sufficient levels.[93]
Figure 2.2: Blood Donations from
1998-2003
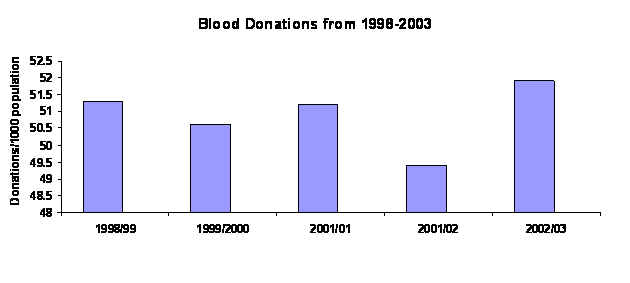 Source: Annual Report 2002-03 Australian Red Cross Blood
Service, p.13.
Source: Annual Report 2002-03 Australian Red Cross Blood
Service, p.13.
2.115
Figure 2.3 shows the total number of blood collections from 1994-95 to
2002-03.
Figure 2.3: Blood collections
1994-95 to 2002-03
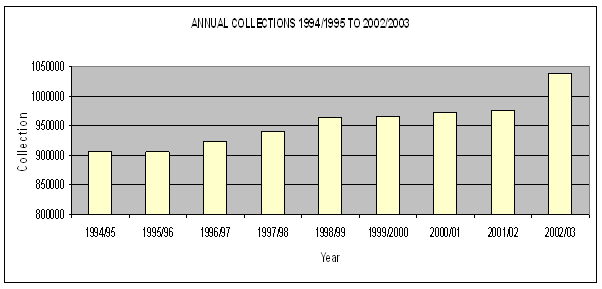
Source:
Annual Report 2002-03 Australian Red Cross Blood Service p.13.
2.116
According to the ARCBS, Australia is in the minority of developed
countries which are able to stay fully self sufficient in fresh blood stocks,
and almost completely self sufficient in plasma products.[94]
This is especially noteworthy as Australia's donors are all voluntary and
totally un-remunerated.
2.117
The mid-1980s saw a considerable tightening of donor eligibility, due to
the advent of HIV/AIDS. This inevitably led to a reduction in the donor pool,
and by 1988 total collections had fallen by 16,000 over the preceding year. It
should be remembered that it was around the time of this decline that the
prospect of surrogate testing, and the attendant reduction in yield, was being
considered in Australia. This reduction in yield was an important concern for
those considering the introduction of the testing.[95]
2.118
Tightening of donor eligibility also had an effect on the supply of
plasma intended for fractionation, although the ARCBS submitted that 'by and
large' the demand for plasma products was still met from within Australia.[96]
The ARCBS notes that certain specialised products, such as Factor VII and
Factor XI, which were required by a small number of patients per year, were
imported.[97]
2.119
Australia's near total self-sufficiency was lauded by the Stephen Review,
which found that:
Under these [largely self-sufficient] circumstances, continuing
high levels of safety and quality should be achievable, as long as careful
national policy measures and strong regulatory oversight are maintained.[98]
2.120
Australia's goal of self sufficiency of blood stocks drew criticism from
the Haemophilia Foundation, which was supportive of the increased use of
recombinant therapies, manufactured overseas, to completely eradicate the risk
of blood-borne virus transmission.[99]
This is discussed further in Chapter 6.
Blood from overseas being used in Australia
2.121
It was submitted by the TBPAG that CSL had 'mixed Australian blood with
blood from several foreign countries for distribution in Australia'.[100]
2.122
The TBPAG refer to an Australian National Audit Office Report relating
to unauthorised processing of foreign-sourced blood plasma by CSL, occurring in
the mid 1990s.[101]
The ANAO report does not conclude that products derived from foreign-sourced
plasma were used in Australia, nor does it conclude that cross-contamination
between foreign and domestic plasma batches occurred.
2.123
In evidence Dr Maher advised that, prior to 1984, CSL blended Australian
and New Zealand plasma for the manufacture of clotting agents where supply was
insufficient from either country. Dr Maher pointed out that similar standards
were applied in each country to the screening of volunteers and donation
testing. Dr Maher then stated:
Apart from the New Zealand example, CSL has never imported or
purchased plasma for the purpose of manufacturing products for therapeutic use
in Australia.[102]
Collection from prison inmates
2.124
The TBPAG raised the Australian Red Cross state divisions' collection of
blood from prison inmates.[103]
The Committee understands from information provided to the Senate that this
practice had ceased by the following approximate dates: New
South Wales, mid 1970s; South Australia, 1975; Western Australia, early
1980s; Victoria, 1983; and Tasmania, 1983.[104]
The global plasma market
2.125
Australia's experience of blood donation stands in contrast to many
other developed nations. In the United States, blood and plasma has for many
years been imported from Europe to supplement the supply required to service
major centres like New York. While paid donation has now been phased out for
fresh blood products, it was a feature of the American blood supply for many
years, and remains an important element in harvesting plasma.[105]
2.126
One critical feature of systems relying on paid donation, compared with
those that are totally voluntary, is the marked increase in the rate of post-transfusion
hepatitis. Indeed, it was this phenomenon which led to the phasing out of paid
blood donation in the U.S, and which played a critical role in Australian
authorities deciding not to proceed with surrogate testing in the mid- to
late-1980s.[106]
2.127
Many nations in Europe are self sufficient, but the UK has struck
difficulty in maintaining supply of plasma, most recently due to the threat of
Creutzfeldt-Jakob Disease being transmitted through the donor pool. As a
result, the UK continues to rely on importation of American (paid) donations.[107]
The special case of haemophiliacs
2.128
Haemophilia is an inherited bleeding disorder which affects about one in
10,000 people. People with haemophilia do not bleed any faster than normal, but
they do bleed longer, due to a deficiency in blood clotting factor. Depending
on severity, haemophiliacs may bleed only after surgery, only after injury or
dental work, or may bleed for no reason at all. In severe cases, bleeding can
occur into muscles and joints, causing extreme pain.
2.129
Haemophilia A is the most common form of haemophilia and is due to a
deficiency of Factor VIII. Haemophilia B is due to a deficiency of Factor IX.
The amount of Factor VIII or Factor IX transfused each year is dependent on the
severity of the haemophilia and frequency of bleeding. Von Willebrand disorder
is another inherited bleeding disorder. Treatment includes infusions of a
clotting factor concentrate that contains von Willebrand factor.
2.130
Until 1964, haemophilia had been treated with blood plasma. In 1964, a
concentration of Factor VIII by freeze thawing of plasma (known as
cryoprecipitate) was developed. From the late 1970s, Factor VIII concentrate
was made by CSL. A Factor IX concentrate called Prothrombinex was also
developed by CSL. Prothrombinex was the major form of treatment of haemophilia
B until it was replaced with a purer Factor IX concentrate (Monofix).[108]
The pooling of thousands of donations of plasma is used to manufacture Factor
VIII and Factor IX concentrates.
2.131
The HFA noted that factor concentrates have revolutionised haemophilia
treatment. They can be made from human blood (called plasma-derived products)
or manufactured using genetically engineered cells that carry a human factor
gene (recombinant products).[109]
Hepatitis C in the haemophilia community
2.132
The HFA reported that following treatment with contaminated blood
clotting factor concentrates, 85 to 90 per cent of people with haemophilia have
been infected with hepatitis C. HFA went on to state that it is likely that up
to 90 per cent of people with haemophilia A and haemophilia B developed NANBH
with their first treatments of non-heat treated factor. There are also more
than 250 people with haemophilia who were infected with HIV and many of these
people are co-infected with HCV.[110]
2.133
The HFA stated that many people with haemophilia in Australia were known
to have hepatitis from the use of blood products and any symptoms they had
'were lived with'. Many did not experience any serious symptoms and the risks
inherent in plasma pooling were balanced against the benefit of the utility of
concentrates. Hepatitis was seen as an unfortunate consequence, but an
acceptable risk of blood products. The HFA concluded that
[I]n reality, people with haemophilia had no choice of whether
or not to use plasma products. When they have severely painful joint or a life
threatening bleeding episode, the decision is clear to use the available
treatment products, even if the treatment may have associated risks.[111]
2.134
The very high prevalence of hepatitis C among people living with
haemophilia can be ascribed to the following three factors:
- the inability to inactivate virus present in plasma and
cryoprecipitate;
- the inability to inactivate NANB hepatitis in pooled plasma
products, prior to the early 1990s; and
- regular use of a number of blood products which were manufactured
from a large number of donations.
2.135
In October/November 1984, CSL adopted a method of preparation of Factor
VIII (used to treat haemophilia A) which allowed for the Factor to be
pasteurised by heating at 60ºC
for 72 hours, thereby destroying some contaminating viruses eg HBV and HIV.
Similar treatment was applied to Factor IX from January 1985.
2.136
The first limited supplies of super heat-treated Factor VIII (80ºC for 72 hours) became available
in January 1990, after reports from Europe of transmission occurring through
Factor heated at the lower temperature.[112]
2.137
Prothrombinex concentrates were heat treated at 60ºC for 72 hours from 1985 onward.
Super heat-treated Factor IX concentrates (heating at 80ºC for 72 hours, shown to inactivate HCV virus) did
not become available in Australia until 1993.[113]
2.138
CSL acknowledged the risks associated with use of Factors VIII and IX
prior to 1989 and 1992, adding that:
[W]ith hindsight...the hepatitis C virus–or Non-A, Non-B hepatitis
as it was known then–was most probably present in every plasma pool throughout
the seventies and the eighties...[i]t is unfortunate that scientific knowledge of
hepatitis C was not sufficient early enough to prevent infection in the
majority of severe haemophilia A and haemophilia B patients treated prior to
the 1990s.[114]
2.139
CSL pointed out that the introduction of heat treatment was initially
controversial. It was argued by some that such practices could lead to an
increase in HAV and HBV positive people who developed inhibitors, a potentially
life-threatening complication characterised by resistance to replacement
therapy. There would also be a reduction in yield. However, the discovery that
HIV was heat sensitive, could be inactivated at 60 degrees, and could otherwise
be transmitted through transfusion, was persuasive.[115]
2.140
CSL went on to remind the Committee that, at the time most heat
treatment was introduced, HCV was still not identified as being a single virus,
and that it was not until the late 1980s that it became clear that 60 degree
heat treatment was insufficient to inactivate the virus which, in 1990, came to
be known as hepatitis C.[116]
2.141
This delay was of concern to the HFA, who submitted that:
There was a considerable delay before Prothrombinex [the Factor
IX based product], heat treated to 80º C, was introduced in mid 1993. This
caused frustration and anxiety for clinicians and patients. Some clinicians
kept their patients on cryoprecipitate to minimise the risk of larger plasma
pools. PTX heat treated to 60º was insufficient to inactivate hepatitis C.[117]
2.142
The HFA also stated that Bio Products Laboratory in the United Kingdom
had increased heat treatment factor VIII to 80 degrees, which prevented
transmission of NANBH, in 1985. However, CSL did not replicate the process
until 1989.[118]
2.143
CSL pointed to the added difficulty of inactivating virus in Factor IX,
saying that fortification against the 80 degree heat treatment necessitated a
substantial reformulation of the product to guard against the occurrence of
thrombosis in recipients.[119]
2.144
The HFA and CSL both stated that there has been no known infection since
additional heat treatment of Factor VIII concentrates in 1989 and Factor IX in
1993.[120]
2.145
The use of recombinant Factor VIII and IX is discussed further in
Chapter 6.
Navigation: Previous Page | Contents | Next Page